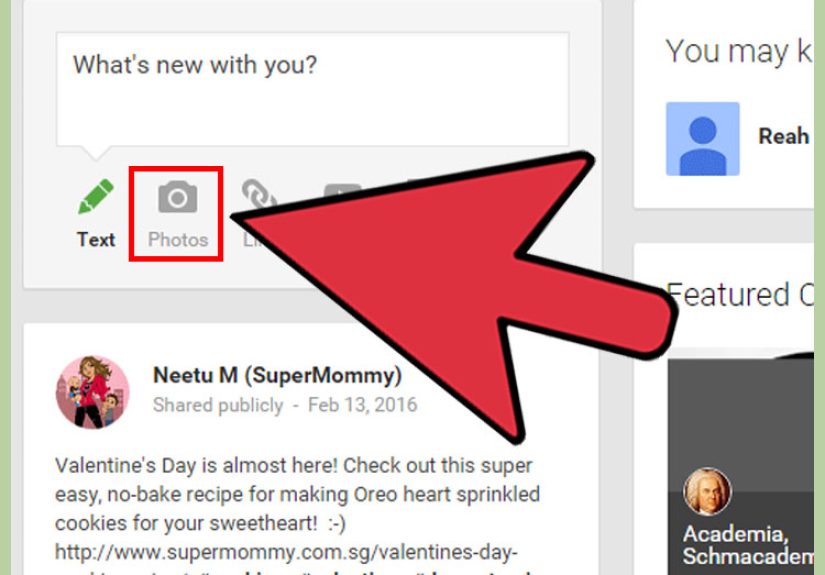




Want to share photos online without blowing up your group chat (or your privacy)? This guide covers...
Having 5 kids teaches a parent fast that perfection is not the point. Real parenting is messy,...
Engagement Chicken is the famous roast chicken that supposedly inspires proposalsbut the real story is even better...
The Remodelista Considered Design Awards opened on June 1, 2018, inviting professional designers, homeowners, renters, and DIY...
Calcium gives bones essential building material, while vitamin D helps the body absorb and use it. This...

Alzheimer's disease involves much more than occasional forgetfulness. This in-depth guide explains 10 essential facts about its...

Castor oil is often promoted online as a quick weight-loss trick, but the science tells a different...

Hey Pandas, Discuss Your Problems is a thoughtful, human-centered look at why sharing struggles matters. From stress...

Cooked a mountain of spaghetti and don’t want it turning into a mushy mystery container? This in-depth...

Calorie counting can help people understand portions, energy intake, and eating patternsbut it is not a magic...